Introduction
Thermodynamics is the study of energy and its interconversions. Chemical Thermodynamics is that portion of thermodynamics that pertains to chemistry and chemical change. You may not have known it, but you began your study of chemical thermodynamics in CHEM 151 (or other first semester general chemistry class) with the study of thermochemistry, which is the study of energy related to physical and chemical change. In CHEM 152, you will continue the study of thermodynamics.
Chemical thermodynamics will give us information about the energy (required or released, depending on whether it is endothermic or exothermic) and direction of chemical reactions. It can help us answer the questions of whether or not a reaction will proceed (direction) and what will be the energy involved. When you studied thermochemistry in your first semester course, you studied the energy piece largely described by the First Law of Thermodynamics). We will review that material as needed as we work through the objectives for chemical thermodynamics, but if you want a more complete review, please feel free to check out my thermochemistry notes from CHEM 151.
The direction piece of the chemical reaction will be described by the Second Law of Thermodynamics, which will likely be new material to you.
Objective 1: Define or explain and give an example of each of the following terms: spontaneous process, state function, system, surroundings. reversible and irreversible processes
The Law of Conservation of Energy (First Law of Thermodynamics)
The First Law of Thermodynamics states that energy cannot be created or destroyed (so it is also known as the the Law of Conservation of Energy). Even though energy is conserved, it can be converted from one type to another. As an example, kinetic energy can be converted to potential or vice versa.
Even though energy can flow from system to surroundings or surroundings to system, The total energy of the universe is a constant. If the system loses energy, it must be gained by the surroundings, and vice versa. (If you’re not sure or don’t recall what I mean when I talk about “system and surroundings”, we’ll review that in a bit.
Mathematically the First Law can be stated as
where
- Δ = greek letter delta, signifies “change in”
- ΔE is change in internal energy
- q = heat
- w = work
Energy used to move an object over some distance is work (w). Heat (q) is the transfer of energy between 2 objects due to a temperature difference. Heat flows from the higher temperature object to the lower temperature object. The primary unit we will use for energy, heat, and work is the joule (J). Since the energy involved in chemical reactions is typically in the thousands of joules per mole of reactant, we will also use kilojoules (kJ).
The First Law of Thermodynamics does not explain why a particular process occurs in a given direction. This is answered by the Second Law, which we will get to a bit later.
System and Surroundings
The system is defined as the collection of matter we are evaluating. Since this is a chemistry class, the system will usually be a chemical reaction.The surroundings is everything in the universe that’s not in the system.
Energy flow between system and surroundings
If energy flows from system to surroundings, ΔE is negative. As is the case with all mathematical deltas, 

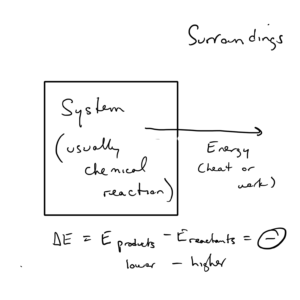
If energy flows from surroundings to system, ΔE is positive. Since , examples could be reactions where the system absorbs heat (positive value of q) or the surroundings does work on the system (positive value of w).
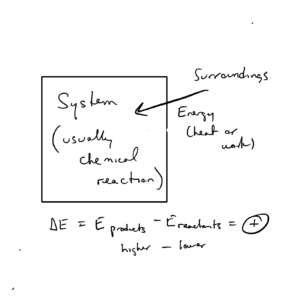
About the positive or negative signs for ΔE
The positive and negative signs are just a way of telling is the direction of energy flow. A negative ΔE means the system is losing energy to the surroundings (flow system to surroundings), while a positive ΔE means the system is gaining energy from the surroundings (flow surroundings to system). This sign convention will be similar for other functions we discuss like:
- ΔH (you likely remember but we will we will review)
- ΔS and ΔG (we will discuss later)
What is important is that you understand the direction of energy flow; you can communicate that either by words describing the direction or by mathematical sign.
Spontaneous Processes
So far we have been reviewing concepts from thermochemistry. The idea of spontaneous processes will be the beginning of our examination of how thermodynamics gives us information of the direction of the reaction — we will go back to review more later as needed.
Spontaneous processes have a natural tendency to occur on their own, in one direction, without any outside intervention. One example is that heat flows from a hotter object to a cooler object. Let’s look at a couple more examples that will be instructive:
- Propane burns to form CO2 and H2O in an exothermic reaction. In the combustion of propane, it reacts with oxygen to produce CO2 , H2O, and heat. So logically speaking, one might say we should be able to put heat into CO2 and H2O and produce propane and oxygen. But we know this is not true. The process is irreversible — it cannot be reversed by simply adding heat.
- Imagine that you have a tub of cool water. You could pour a bucket of boiling water into the tub , raising the temperature of the water so you had a tub of warm water. One might think logically, then, that if you started with a tub of warm water, you should be able to scoop a bucket of boiling water out of it. leaving a tub of cool water behind! Of course, that is ridiculous! Dumping a bucket of boiling water into cool water to make warm water is irreversible – we can’t undo it by exactly reversing the process.
In a reversible process the system changes in such a way that the system and surroundings can be put back in their original states by exactly reversing the process. Irreversible processes cannot be undone by exactly reversing the change to the system. The examples above cannot be undone by exactly reversing the system. They are irreversible. You could reverse process 2 above by intervening – scoop out a bucket of water and heat it to boiling – but this is outside intervention and is not exactly reversing the process.
Spontaneous processes – remember, these are ones that have a natural tendency to occur on their own, without any outside intervention – are irreversible. Processes that are spontaneous in one direction are not spontaneous in the other direction.
Two functions contribute to spontaneity – one of them (enthalpy) should be familiar to you from previous study, the other (entropy) is likely new:
- The enthalpy, H — Is the reaction endothermic or exothermic? Exothermic reactions are more likely to be spontaneous, but it is not a guarantee.
- The entropy, S — we will discuss this soon.
Enthalpy (H) – review
Enthalpy is defined as:
H = E + PV
where H is enthalpy, E is internal energy, P is pressure, and V is volume.
Enthalpy is useful since, at constant pressure (many chemical reactions occur at constant pressure):
q=ΔH
where q=heat and ΔH is change in enthalpy.
OpenStax section 5.3 (read the section just below the mountain picture) shows mathematically why this is the case.
What is convenient about q=ΔH at constant pressure?
Heat flow is relatively easy to measure. Since chemical reactions are often run at constant pressure, we can measure the heat and know the enthalpy change, which is quite useful since enthalpy is a state function (if you don’t remember state functions, we will review that next). We can do this without having to measure the work, which is not measured as easily.
In a chemical reaction – the flow of heat into or out of the reaction system is the change in enthalpy of the system and is called the enthalpy change of reaction or the heat of reaction. Note H is enthalpy. It is not heat. ΔH is just equal to the heat gained (required) or lost (produced) by the reaction, so we say “heat of reaction”.
Heat of reaction (ΔH)

If ΔH is positive (Hproducts > Hreactants)
Heat from the surroundings is absorbed by the system, meaning the reaction requires energy to proceed. These reactions are called endothermic reactions.
If ΔH is negative (Hproducts < Hreactants)
Heat is lost by system to the surroundings, meaning the reaction produces energy as heat. These reactions are called exothermic reactions.
Thermodynamic Properties and State Functions
State functions are path independent (not path dependent). The value of a state function does not depend on how the substance got to that state. Examples of state functions are:
- temperature (T)
- kinetic energy
- potential energy
- internal energy (E)
- pressure (P)
- volume (V)
- enthalpy (H)
- entropy (S)
Functions that are path dependent are NOT state functions. Their value does depend on path. Heat and work are examples.
The idea of state functions is somewhat abstract; OpenStax Figure 5.20 and the text around it gives a nice illustrative example.
Why is the concept of state functions useful?
The concept of state functions is useful because it allows us to calculate changes in. thermodynamic properties more easily. Any path that takes us from reactants to products (even if it is not the actual path) will give the change in that property for the reaction. As examples:
- We can calculate ΔH using standard heats of formation
- Calculate ΔH using Hess’s Law
Next, we will review standard heats of formation.
Review: Standard Enthalpy of Formation or Standard Heat of Formation (ΔH0f)
The enthalpy change associated with the formation of a compound from its constituent elements is called the enthalpy of formation (ΔHf). The standard enthalpy of formation (ΔH°f) of a compound is the change in enthalpy for the reaction that forms one mole of the compound from its elements with all substances in their standard states (superscript o indicates standard state conditions).
What is meant by standard states?
In thermodynamics, standard states mean the following:
- T = 25 °C
- For gases, pressure = 1 atm
- For aqueous solutions (aq), concentration = 1 M
- liquids and solids are pure
- elements are in their form they typically exist at in standard conditions. For example:
- Iron: Fe(s) is standard. Fe(l) is not, since iron is a solid at standard state.
- oxygen: O2 (g) is standard state, as oxygen exists as a diatomic gas at standard state. O(g), O(l), O2(l) are not.
Standard heats of formation are tabulated in a variety of places. The values can vary slightly depending on the source, so if you get an answer that is close but slightly different that is likely the reason.
ΔH0f is ΔH for a reaction forming one mole of a compound from its elements with all substances in their standard states. ΔH0f values are listed in Appendix G in OpenStax. You can find them via Google search as well.
Using Standard Enthalpies of Formation: The standard enthalpy change for any reaction (ΔH°) can be calculated from the standard enthalpies of formation of the reactants and products using:

Standard Heat of Formation example
Using standard enthalpies of formation, calculate the heat of reaction for:

Given the following data (which could be looked up):
| Compound | ΔHf° (kJ/mol) |
| CO2 (g) | -393.5 |
| H2O (l) | -285.83 |
| C3H8 (g) | -103.85 |
Notice O2 (g) is not given. As an element in its standard state, its ΔHf° value is zero. It takes no energy to form O2 (g) from O2 (g). ΔHf° values are zero for all elements in their standard states.
![\Delta H=\left[ 3\text{ mol}\left( -393.5\frac{kJ}{mol} \right)+4\text{ mol}\left( -285.83\frac{kJ}{mol} \right) \right]-\left[ 1\text{ mol}\left( -103.85\frac{kJ}{mol} \right)+5\text{ mol}\left( 0\frac{kJ}{mol} \right) \right]](https://s0.wp.com/latex.php?latex=%5CDelta+H%3D%5Cleft%5B+3%5Ctext%7B+mol%7D%5Cleft%28+-393.5%5Cfrac%7BkJ%7D%7Bmol%7D+%5Cright%29%2B4%5Ctext%7B+mol%7D%5Cleft%28+-285.83%5Cfrac%7BkJ%7D%7Bmol%7D+%5Cright%29+%5Cright%5D-%5Cleft%5B+1%5Ctext%7B+mol%7D%5Cleft%28+-103.85%5Cfrac%7BkJ%7D%7Bmol%7D+%5Cright%29%2B5%5Ctext%7B+mol%7D%5Cleft%28+0%5Cfrac%7BkJ%7D%7Bmol%7D+%5Cright%29+%5Cright%5D&bg=ffffff&fg=000&s=0&c=20201002)

This reaction is quite exothermic, which is to be expected for the combustion of propane!
Standard Heat of Formation practice
Using the standard heats of formation from Appendix G in OpenStax , calculate ΔH° for the following reaction:

Make sure you understand the above calculation! Not only will we use it for ΔH calculations, but we will also use it for ΔS and ΔG calculations. You may have noticed that in Appendix G in OpenStax , there are columns for ΔGf° and S°. The calculations for ΔS and ΔG will use these columns and work similarly.
As mentioned earlier, exothermic processes are more likely to be spontaneous than endothermic ones. But to determine spontaneity we must also consider entropy (S). We will look at that function next.
Objective 2: Explain how entropy is related to randomness or disorder, positional probability, and the number of microstates available to the system.
Objective 4: Predict the relative number of microstates available to a system under varying conditions, or using your understanding of the relative number of microstates available to systems predict whether entropy of a system increases or decreases.
Entropy can be described as a measure of molecular randomness or disorder. The higher the degree of disorder, the greater the entropy. In general, spontaneous processes lead to an increase in entropy (positive value of ΔS) . The symbol S is used for entropy and its units are J/K.

Entropy is closely tied to probability. Let’s look at how with an example:
Imagine you have a giant drum with a million coins on it. If you beat the drum with enough force, the coins will bounce around. When you stop playing the drum, how likely would it be that you would get all heads or all tails?
The answer is, of course, very unlikely (we will get to how just unlikely in a moment). Before we look at the case of a million coins, lets look at something more manageable — four coins.
If four coins are bouncing around on the drum, the possible outcomes are this:
4 heads – HHHH (1 way to get this)
3 heads, 1 tails – HHHT, HHTH, HTHH, THHH (4 ways)
2 heads, 2 tails – HHTT, HTHT, HTTH, THHT, THTH, TTHH (6 ways)
1 heads, 3 tails – HTTT, THTT, TTHT, TTTH (4 ways)
4 tails – TTTT (1 way)
The most likely outcome is 2 heads, two tails, and the further one gets to this 50/50 split, the less likely a particular outcome. There are 16 possible outcomes (the 1+4+6+4+1 ways above) and only one of them is all heads, giving that a 1/16 probability.
The probability of all heads with 4 coins is

The probability of all heads with a million coins is

Let’s look at an example now that is more relevant to a chemistry class than my ridiculous example of a million coins on a giant drum….
Imagine you have 1 mole of an ideal gas in a flask system like the figure below (Figure 16.4 OpenStax)
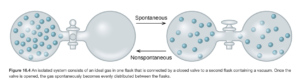
In an ideal gas, each individual molecule or atom is so far apart from the next one compared to its size that the individual molecules do not affect each other in any way, and each molecule is free to move anywhere it wants without interference, just as if it were the only molecule on the containers. If we start with the diagram on the left (all the gas in the left flask) and open the valve separating the flask, you would expect gas to go to the right, and settle with equal amounts of gas on both sides. This will happen spontaneously. But if we started with the diagram on the right, you would not expect all the gas to move to the left (that is nonspontaneous).
This is for exactly the same reason as the coins on the drum.. All the gas could end up on the left (think of all the gas in the left as heads and gas on the right flask as tails), but the probability that it will is 
The spontaneous case is the one on the right (50/50 split) because there are so many more ways it can occur. This is referred to as positional probability.
Positional Probability and Microstates
The coins on the drum and the gas in the container illustrate positional probability.
Positional probability depends on how many ways something can happen. There are many more ways the gas can be split close to evenly on both sides than all on one side. Entropy is proportional to the number of ways or the positional probability.
At the molecular level, entropy can be described in terms of the possible number of different arrangements of particle positions and energies, called microstates. Each thermodynamic state has a characteristic number of microstates (W) which are related to entropy (S) by the Boltzmann equation S = k lnW, where k is Boltzmann’s constant. The more microstates the system has, the greater its entropy. Entropy is thus a measure of how many microstates are associated with a particular macroscopic state. Any change in the system that increases the number of microstates gives a positive value of ΔS and vice versa. The gas will spontaneously go from the state in the flask system on the left (only 1 microstate, low S) to the state in the flask system on the right (many microstates, high S), because it is much more probable.
Objective 3: Predict whether a process is spontaneous.
Objective 5: Predict the sign of the entropy change for a given process, phase change, or chemical reaction.
Objective 6: Calculate ΔS° for a chemical reaction using the tabulated S° values of the reactants and products and a balanced equation.
Objective 7: State and explain the second law of thermodynamics in terms of entropy. Understand the relationship of the entropy change in the system, surroundings, and universe to the second law.
Objective 8:Explain the temperature dependence of entropy and predict how spontaneity can be affected by increasing or decreasing temperature.
Effect of Phase of Matter on Entropy
For the same substance:

As a substance goes from solid, to liquid to gas, disorder (and entropy) increases due to the increasing number of microstates available.
This means for example, that solid H2O (ice) is lower in entropy than liquid H2O, and liquid H2O is lower in entropy than gaseous H2O (steam). It does not mean that all solids are lower in entropy than all liquids, etc.
Given that, answer the following:
The Second Law of Thermodynamics
According to the Second law of Thermodynamics, the total entropy of the universe(

For a system (in this class, most often a chemical reaction),
The direction of a spontaneous change is always determined by the sign of the total entropy change:
- The reaction or process is spontaneous if
- The reaction or process is nonspontaneous if
(and the reverse reaction is spontaneous)
- The reaction or process is at equilibrium if
.
Looking at
| ΔSsystem | ΔSsurroundings | ΔSuniverse | spontaneous? |
| + | + | + | yes |
| – | – | – | no |
| + | – | ? (depends on which is bigger) | maybe |
| – | + | ? | maybe |
Relationship of ΔS surroundings to enthalpy change of the system
At first glance, it looks like it might be difficult to determine ΔS of the surroundings, as it contains everything other than the system. But we can relate it to the enthalpy change of the system, or specifically whether the system is exothermic or endothermic. Let’s consider an exothermic system, which by definition releases heat to the surroundings:
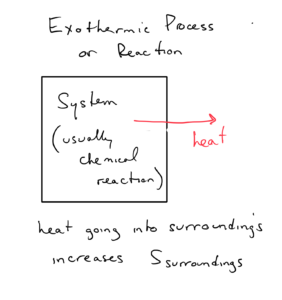
As the exothermic process releases heat to the surroundings, it increases the temperature of the surroundings, which increases the kinetic energy of the molecules in the surroundings. This increased energy of motion results in more microstates, higher positional probability, and higher entropy of the surroundings.
An exothermic system means a positive ΔSsurroundings . The opposite is also true. An endothermic system means a negative ΔSsurroundings . The mathematical relationship is

where T is temperature in Kelvin. This is for an isothermal process (temperature of the system remains constant).
You may recall I stated earlier (in Objective 1) that exothermic reactions were more likely to be spontaneous — the reason is that they increase the entropy of the surroundings. The entropy of the system still needs to be considered, though. Next, we will look at how we can make qualitative predictions about entropy changes for systems.
Making Qualitative Predictions About ΔS
Before looking at this — a little about notation.
SYSTEM IS DEFAULT. What do I mean by this? If we are looking at the surroundings or universe with any function (ΔS, ΔH, or later ΔG), it will be denoted:
ΔSsurr means ΔSsurroundings
ΔSuniv means ΔSuniverse.
If nothing is stated, it is assumed we are looking at the system.
ΔS means ΔSsystem
I will assume you are doing the same.
Now, back to Making Qualitative Predictions About ΔS
Temperature effects
In most cases an increase in temperature means the number of microstates (and thus entropy). A temperature increases means ΔS is positive.
Volume
In most cases an increase in volume means the number of microstates (and thus entropy). A volume increases means ΔS is positive.
Phase
As discussed earlier, for the same substance:
As a substance goes from solid, to liquid to gas, disorder (and entropy) increases due to the increasing number of microstates available.
Processes that will generally increase entropy (positive ΔS)
In general, entropy will increase when:
- Liquids or solutions are formed from solids
- Gases are formed from solids or liquids
- Dissolving ionic solids in water. Example:
- The number of gas molecules increases. Example:
.

If a reaction goes from fewer moles of gas to more moles of gas, ΔS is almost always positive. If a reaction goes from more moles of gas to fewer moles of gas, ΔS is almost always negative.
Complexity of gaseous molecules
For reactions with the same number of moles of gas on the reactant and product side, it is useful to look at the identity of the gas molecules. Let’s consider gas molecules with 2 atoms:
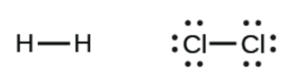
As they are heated, the molecules can vibrate – in molecular vibrations, the atoms move relative to each other.
Gas molecules that have more atoms (such as the ones below)
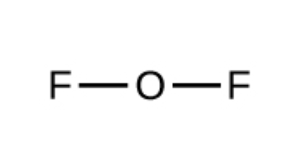
can vibrate in more ways, leading to more microstates and higher entropy.
Making Qualitative Predictions About ΔS – Practice
Spontaneity of some reactions or processes can depends on temperature
Earlier we looked at four possibilities when using the Second Law and entropies of the system and surroundings to predict spontaneity:
| ΔSsystem | ΔSsurroundings | ΔSuniverse | spontaneous? |
| + | + | + | yes |
| – | – | – | no |
| + | – | ? (depends on which is bigger) | maybe |
| – | + | ? | maybe |
The last two rows (that may or may not be positive) depend on temperature. A notable example of this are phase changes. The boiling of a liquid is spontaneous above the boiling point but nonspontaneous below the boiling point. The condensation of a gas is spontaneous below the boiling point. Let’s look at this a little closer using water as an example:
We can describe our system as

ΔHsystem is positive since boiling is endothermic
ΔSsurr is negative since ΔHsystem is positive
ΔSsystem is positive since going from a liquid to a gas
Therefore, the spontaneity depends on which magnitude or absolute value is bigger: ΔSsurr or ΔSsystem .
- At temperatures above 100°C, ΔSuniv is positive and boiling is spontaneous.
- At temperatures below 100°C, ΔSuniv is negative and boiling is not spontaneous (the opposite, condensation, is).
- At a temperature 100°C, ΔSuniv is zero and liquid and gas are at equilibrium
Third Law of Thermodynamics
The entropy of a pure crystalline substance at absolute zero is 0. For an entropy of zero, there must be no disorder. This means a perfect crystal – every atom or molecule in place at temperature of absolute zero (where there is no atomic motion).
The importance of the third law to us is that is sets a zero point for entropy – and standard entropies (S°) can be measured and tabulated relative to that. This makes it easy to calculate entropy changes for reactions similarly to the method used with heat of formation, since entropy is also a state function
Tabulated Standard Entropy Values
S° is standard entropy at 298 K and 1 atm. These values are always greater than zero. S° values are tabulated in Appendix G in OpenStax. You can find them via Google search as well.
S° values are different from ΔH° in some ways
- S° values are absolute: based on a reference point of zero for a perfect crystalline solid at zero K (the 3rd law).
- Standard molar entropies of elements are not
- It tends to increase with the number of atoms in the formula of the substance.
- It tends to increase with increase in molar mass
- The units are

Using Tabulated Standard Entropy Values – example
Using standard entropy values, calculate ΔS° for the reaction

![\Delta {{S}^{0}}=2{{S}^{0}}_{N{{H}_{3}}}-\left[ {{S}^{0}}_{{{N}_{2}}}+{{S}^{0}}_{{{N}_{2}}} \right]](https://s0.wp.com/latex.php?latex=%5CDelta+%7B%7BS%7D%5E%7B0%7D%7D%3D2%7B%7BS%7D%5E%7B0%7D%7D_%7BN%7B%7BH%7D_%7B3%7D%7D%7D-%5Cleft%5B+%7B%7BS%7D%5E%7B0%7D%7D_%7B%7B%7BN%7D_%7B2%7D%7D%7D%2B%7B%7BS%7D%5E%7B0%7D%7D_%7B%7B%7BN%7D_%7B2%7D%7D%7D+%5Cright%5D&bg=ffffff&fg=000&s=0&c=20201002)
![\text{ }\!\!~\!\!\text{ }\!\!~\!\!\text{ }\!\!~\!\!\text{ }\!\!~\!\!\text{ }\Delta {{\text{S}}^{\text{0}}}\text{= }\!\!~\!\!\text{ 2 }\!\!~\!\!\text{ mol}\left( \text{192.5 }\!\!~\!\!\text{ }\frac{\text{J}}{\text{mol }\!\!\cdot\!\!\text{ K}} \right)\text{ }\!\!~\!\!\text{ --}\left[ \text{1 }\!\!~\!\!\text{ mol}\left( \text{191.5 }\!\!~\!\!\text{ }\frac{\text{J}}{\text{mol }\!\!\cdot\!\!\text{ K}} \right)\text{ }\!\!~\!\!\text{ + }\!\!~\!\!\text{ }\!\!~\!\!\text{ 3mol}\left( \text{130.6}\frac{\text{J}}{\text{mol }\!\!\cdot\!\!\text{ K}} \right) \right]\text{ }\!\!~\!\!\text{ }\!\!~\!\!\text{ }](https://s0.wp.com/latex.php?latex=%5Ctext%7B+%7D%5C%21%5C%21%7E%5C%21%5C%21%5Ctext%7B+%7D%5C%21%5C%21%7E%5C%21%5C%21%5Ctext%7B+%7D%5C%21%5C%21%7E%5C%21%5C%21%5Ctext%7B+%7D%5C%21%5C%21%7E%5C%21%5C%21%5Ctext%7B+%7D%5CDelta+%7B%7B%5Ctext%7BS%7D%7D%5E%7B%5Ctext%7B0%7D%7D%7D%5Ctext%7B%3D+%7D%5C%21%5C%21%7E%5C%21%5C%21%5Ctext%7B+2+%7D%5C%21%5C%21%7E%5C%21%5C%21%5Ctext%7B+mol%7D%5Cleft%28+%5Ctext%7B192.5+%7D%5C%21%5C%21%7E%5C%21%5C%21%5Ctext%7B+%7D%5Cfrac%7B%5Ctext%7BJ%7D%7D%7B%5Ctext%7Bmol+%7D%5C%21%5C%21%5Ccdot%5C%21%5C%21%5Ctext%7B+K%7D%7D+%5Cright%29%5Ctext%7B+%7D%5C%21%5C%21%7E%5C%21%5C%21%5Ctext%7B+--%7D%5Cleft%5B+%5Ctext%7B1+%7D%5C%21%5C%21%7E%5C%21%5C%21%5Ctext%7B+mol%7D%5Cleft%28+%5Ctext%7B191.5+%7D%5C%21%5C%21%7E%5C%21%5C%21%5Ctext%7B+%7D%5Cfrac%7B%5Ctext%7BJ%7D%7D%7B%5Ctext%7Bmol+%7D%5C%21%5C%21%5Ccdot%5C%21%5C%21%5Ctext%7B+K%7D%7D+%5Cright%29%5Ctext%7B+%7D%5C%21%5C%21%7E%5C%21%5C%21%5Ctext%7B+%2B+%7D%5C%21%5C%21%7E%5C%21%5C%21%5Ctext%7B+%7D%5C%21%5C%21%7E%5C%21%5C%21%5Ctext%7B+3mol%7D%5Cleft%28+%5Ctext%7B130.6%7D%5Cfrac%7B%5Ctext%7BJ%7D%7D%7B%5Ctext%7Bmol+%7D%5C%21%5C%21%5Ccdot%5C%21%5C%21%5Ctext%7B+K%7D%7D+%5Cright%29+%5Cright%5D%5Ctext%7B+%7D%5C%21%5C%21%7E%5C%21%5C%21%5Ctext%7B+%7D%5C%21%5C%21%7E%5C%21%5C%21%5Ctext%7B+%7D&bg=ffffff&fg=000&s=0&c=20201002)

Using Tabulated Standard Entropy Values – practice
Consider the following reaction: N2(g) + 3H2(g) → 2NH3(g)
Given that
- ∆So = -198.2
- So(NH3) = 192.5
- So (N2) = 191.5


calculate the entropy So of H2(g).
Objective 10: Define free energy and relate it to spontaneity.
Gibbs Free Energy (G) is a state function where G=H-TS
Gibbs Free Energy is a function of convenience. As you will see in a moment, it will allow us to determine whether or not a reaction will be spontaneous by considering the system only. Since G=H-TS, at constant temperature and pressure we can say

If we divide both sides of this equation by -ΔT:

which can be simplified to:

cancelling the T on the right and recalling that 

or

Since kelvin T is always positive, this means that ΔGsystem and ΔSuniv are always opposite in sign.
Therefore,
- The reaction or process is spontaneous if
or ΔG is negative
- The reaction or process is nonspontaneous if
- The reaction or process is at equilibrium if
The same four possibilities we saw when looking at the second law can be expressed in terms of ΔG. You might recall, in the instances where it might or might not be spontaneous, it depended on the temperature. These four possibilities are listed in Figure 16.12 on OpenStax, which is shown below:
Objective 10 Practice
For each reaction below, indicate whether it is always spontaneous, never spontaneous, spontaneous at high T, or spontaneous at low T). Hint: you will have to figure out whether ΔS is positive or negative first.




Objective 9: Given any two of the following variables, ΔS, ΔH or T calculate the third variable.
Objective 11: Given ΔH, ΔS, and T, calculate the change in Gibbs Free Energy and then predict whether a process will be spontaneous.
Objective 12: Define and write the equation for the standard free energy of formation, ΔGf° of a substance.
Objective 13: Calculate ΔG°, ΔH° and ΔS° for a reaction from tabulated data.
Objective 14:Given temperature and any two of the following variables, ΔS, ΔH or ΔG, calculate the third variable
Objective 19: Predict how ΔG° will change with temperature given ΔH° and ΔS°.
Standard Free Energy Change (ΔG°)
Standard Free Energy Change (ΔG°) is simply the free energy change under standard conditions.
Recall, standard conditions means:
- T = 25 °C
- For gases, pressure = 1 atm
- For aqueous solutions (aq), concentration = 1 M
- liquids and solids are pure
- elements are in their form they typically exist at in standard conditions. For example:
- Iron: Fe(s) is standard. Fe(l) is not, since iron is a solid at standard state.
- oxygen: O2 (g) is standard state, as oxygen exists as a diatomic gas at standard state. O(g), O(l), O2(l) are not.
It indicates whether a reaction will proceed spontaneously. Additionally, it can give us an idea of whether the reactants or products will be favored in an equilibrium process. The more negative ΔG° is, the further right the process will go to reach equilibrium, and the larger the K. Unlike ΔH°, which can be experimentally determined using calorimetry, ΔG° must be calculated.
Calculation of Standard Free Energy Change (ΔG°)
ΔG° can be calculated by several methods:
ΔG° can be calculated using ΔG°=ΔH°-TΔS°
ΔG° can be calculated by the equation
Note that is the same equation as the earlier 
In the equation 
Make sure to consider units when solving these problems. ΔG° and ΔH° are typically given in kJ, while ΔS° is normally in J/K. You must work either all in J or all in kJ. The most common error on these problems is to ignore units and simply add the numbers, leading to incorrect results.
ΔG° can be calculated using tabulated values for the standard free energies of formation (ΔG°f)
This is a similar calculation to ΔH° from ΔH°f values or ΔS° from S° values. ΔG°f is ΔG for a reaction forming one mole of a compound from its elements with all substances in their standard states. ΔG°f values are tabulated in your textbook in Appendix G in OpenStax. ΔG° can also be calculated using Hess’s Law
Finally, ΔG° can be calculated by manipulating known equations as in Hess’s Law problems for ΔH°. If you know ΔHo(or ΔGoor ΔSo) for 2 or more reactions that add to another reaction, you can add the ΔHo(or ΔGoor ΔSo) values for each of the reactions to get the ΔHo(or ΔGoor ΔSo) for the overall reaction. For a review of Hess’s Law, see Objective 8 in my thermochemistry notes.
Objective 9, 11-14, 19 Practice
In these practice questions, you will practice some of the different problem solving strategies and equations for calculating ΔG° discussed above. As part of these problems, you may also have to use standard ΔGf° and S° values to calculate ΔG° and ΔS°.
Calculate the temperature at which the following reaction will occur spontaneously:

Given ΔH° = 98.8 kJ; ΔS° = 141.5 J/K
Calculate ΔG° for the melting of ice -10°C. ΔH°= 6.033 kJ/mol and ΔS°=22.1 J/mol K
Calculate the boiling point of a liquid using its heat of vaporization ΔH° = 40.7 kJ/mol the entropy change for the phase change ΔS° = 109 J/mol⋅K.
Hint: Recognize this change as an equilibrium system; ΔG = 0
Using the data below to first calculate ΔH° and ΔS°, calculate ΔG° for the following reaction at 25°C:

| Compound | ΔHºf (kJ/mol) | Sº (J/mol·K) |
| CH4(g) | -75 | 186 |
| O2(g) | 0 | 205 |
| CO2(g) | -393.5 | 214 |
| H2O(g) | -242 | 189 |
Using the following tabulated values for the standard free energies of formation, calculate ΔG° for the reaction:
ΔGºf for CH4 = -50.8 kJ/mol
ΔGºf for CO2 = -394.4 kJ/mol
ΔGºf for H2O = -228.57 kJ/mol
Comparing the answers to the previous two practice problems, are the results to be expected? They should be, given that it is the same reaction.
Given the following data


Calculate ΔG° for the reaction

Objective 15: Explain how ΔG changes with a change in the concentration or partial pressure of reactants or products.
Objective 16: Calculate ΔG given the concentration or partial pressures of reactants and products.
Objective 17: Explain equilibrium in terms of minimum free energy and show how the value of K is related to free energy.
Objective 18: Calculate ΔG° from K and vice-versa.
So far, we have looked at free energy change primarily under standard conditions:
- T = 25 °C
- For gases, pressure = 1 atm
- For aqueous solutions (aq), concentration = 1 M
At non-standard conditions, free energy ΔG will be different from the standard free energy ΔG°. Let’s first consider the effect of pressure in gases:
Slarger volume > Ssmaller volume as a gas at larger volume has more microstates.
Since PV=nRT, pressure and volume are inversely proportional. Larger volume is lower pressure. Therefore, Slower pressure > Shigher pressure.
Since G=H-TS, a higher S means a lower G, so Glower pressure < Ghigher pressure.
What this tells us is that changing pressure changes G, so at pressures other than 1 atm G will change.
Since PV=nRT, 

Glower concentration < Ghigher concentration
and molarities deviating from the standard molarity of 1M will have different free energies. So how do we determine G and ΔG in nonstandard conditions? That’s next.
ΔG for non standard conditions
The following equation relates standard and nonstandard ΔG:
Where
- ΔG = free energy change (nonstandard conditions)
- ΔG° = free energy change (standard conditions)
- R = 8.3145 J/ mol·K
- T = Kelvin temperature
- Q = reaction quotient
All of these variables should be familiar to you except perhaps Q, the reaction quotient. You likely learned about Q in your study of equilibrium in CHEM 151 or other first semester course. Let’s review it.
Review from CHEM 151: Reaction Quotient Q
For the reaction 
![K=\frac{{{[\text{N}{{\text{H}}_{3}}]}^{2}}}{[{{\text{N}}_{2}}]{{[{{\text{H}}_{2}}]}^{3}}}](https://s0.wp.com/latex.php?latex=K%3D%5Cfrac%7B%7B%7B%5B%5Ctext%7BN%7D%7B%7B%5Ctext%7BH%7D%7D_%7B3%7D%7D%5D%7D%5E%7B2%7D%7D%7D%7B%5B%7B%7B%5Ctext%7BN%7D%7D_%7B2%7D%7D%5D%7B%7B%5B%7B%7B%5Ctext%7BH%7D%7D_%7B2%7D%7D%5D%7D%5E%7B3%7D%7D%7D&bg=ffffff&fg=000&s=0&c=20201002)
Since it is a gas-phase reaction, we could also write an equilibrium constant Kp using partial pressures:

In equilibrium constants, the concentrations and partial pressures are equilibrium concentrations and equilibrium partial pressures.
We can also write reaction quotients Q and Qp
![Q=\frac{{{[\text{N}{{\text{H}}_{3}}]}^{2}}}{[{{\text{N}}_{2}}]{{[{{\text{H}}_{2}}]}^{3}}}](https://s0.wp.com/latex.php?latex=Q%3D%5Cfrac%7B%7B%7B%5B%5Ctext%7BN%7D%7B%7B%5Ctext%7BH%7D%7D_%7B3%7D%7D%5D%7D%5E%7B2%7D%7D%7D%7B%5B%7B%7B%5Ctext%7BN%7D%7D_%7B2%7D%7D%5D%7B%7B%5B%7B%7B%5Ctext%7BH%7D%7D_%7B2%7D%7D%5D%7D%5E%7B3%7D%7D%7D&bg=ffffff&fg=000&s=0&c=20201002)

While Q and K look the same, the difference is that in Q, the concentrations or partial pressures may or may not be at equilibrium. Q will tell whether or not the system is at equilibrium (if Q=K it is, and if 
- Q<K : the reaction will shift right to reach equilibrium
- Q>K : the reaction will shift left to reach equilibrium
- Q=K : the reaction is at equilibrium
Objective 16 example
For the reaction

ΔG° = -29 kJ/mol ( make sure you know how to calculate this !! – it will not always be given). For the various ways to calculate ΔG° , see the previous objectives and practice problems.
If CO has an initial partial pressure of 5.0 atm, and H2 has an initial partial pressure of 3.0 atm, calculate ΔG at a temperature of 298 K.
For this reaction,

Using 

yielding ΔG=-38431 J, or (rounding for the sig fig subtraction rule) ΔG=-38 kJ
Significance of the example’s result and connection between free energy and equilibrium
In the example above, for the reaction
the standard free energy change ΔG° = -29 kJ. This indicates the reaction is spontaneous, and will proceed to the right. Since it is a standard ΔG, -29kJ is the free energy change when both partial pressures are at the standard condition of 1 atm.
In the non standard case, the pressures of both CO and hydrogen were increased (to 3.0 and 5.0 atm) This is, in essence, adding CO and hydrogen. LeChatlier’s principle tells us that should shift the reaction right. The more negative free energy under these conditions (-38 kJ vs -29 kJ) says the reaction is more spontaneous than at standard conditions, or a shift to the right compared to standard. This is not coincidence.
Free energy is related to equilibrium. As long as ΔG is negative, the reaction will proceed spontaneously. Therefore, the system will proceed to the lowest possible free energy. In CHEM 151, you learned from studying the reaction quotient Q that if Q ≠ K the reaction will proceed (spontaneously) toward equilibrium. The shifting left or right, or spontaneity, will continue until equilibrium is reached (learned in CHEM 151) or until free energy can no longer be decreased (learned in this unit). This will be at the same point. Therefore, equilibrium represents the lowest free energy available to the system.
Relationship between ΔG° and K
We learned earlier that
At equilibrium, Q=K and ΔG = 0, therefore
Even though this equation is derived using equilibrium, it is valid whether or not the system is at equilibrium. If we know ΔG° for any reaction, we can calculate its equilibrium constant K. Conversely, If we know K for any reaction, we can calculate ΔG°.
This will be true even for reactions that we do not consider equilibrium, such as the combustion of methane in the next example.
Relationship between ΔG° and K example
For the reaction 
Using 

Plugging in gives


Solving for K,

Note – if your calculator overflows, it is acceptable to to report

This very large value of K indicates the reaction “equilibrium” lies completely to the right, which we would expect for a one-way reaction that is spontaneous.
The following practice question will let you practice with the relationship between ΔG , ΔG° , Q and K with more reasonable equilibrium reactions. In these, remember that K can be any of the special instances of K we have discussed this semester, including
- Kw for the dissociation of water
- Ka for a weak acid dissociation
- Kb for w weak base ionization
- Ksp for a slightly soluble salt.
For the practice below, K is Kw.
Relationship between ΔG° and K practice
Calculate ΔG for the dissociation of water when [H+]0 = 1.0 X 10-5 M and [OH–]0 = 1.0 X 10-9 M
Calculate ΔG for the dissociation of water when [H+]0 = 1.0 M and [OH–]0 = 1.0 X 10-9 M
Objective 20: Explain the relationship between the maximum work obtainable and the free energy change.
Objective 21: Explain the relationship between kinetics and thermodynamics.
Free Energy and Work
The free energy change for a reaction determines how much work can be done with the reaction. The maximum possible work obtainable form a process at constant T and P is equal to the change in free energy – we can think of the free energy as the difference in the energy produced and the energy lost to the surroundings.
ΔG = wmax
A negative value for ΔG means the energy is free to do useful work, while a positive ΔG means the energy is the minimum amount of work that must be expended to make the process occur.
Actual processes such as engines are never 100% efficient, so the actual work done is always less that the maximum. This work is changed to heat in surroundings, increasing Suniv.
Kinetics and Thermodynamics
Earlier in this course, you learned that chemical kinetics tells how fast a a reaction or process will occur — the reaction rate. Thermodynamics tells us whether a reaction or process will occur- spontaneous ones will, non spontaneous ones will not.
Both kinetics and thermodynamics address equilibrium. In kinetics, equilibrium occurs when the forward and reverse reaction rates are equal. In thermodynamics, equilibrium occurs at the lowest free energy. These are the same equilibrium!